Build and simulate your own Model
user_model.RmdIn this article we will assume that you have a reaction network model
that you want to work on and that you have written the model in the
SBtab format. It may be a Google spreadsheet, an open document
spreadsheet (.ods), an MS Excel file, a file for the
“Numbers” application on an Apple device or a .gnumeric
file.
Firstly, you need to build your model, i.e., translate your reaction network model from the SBtab format into machine code that can be used for simulations in UQSA.
Build your model
There are three steps we need to make to build the model:
| Steps | file (out) | tl;dr | |
|---|---|---|---|
| 1 | convert SBtab to ODE | [optional] | biology => math |
| 2 | convert the ODE to C code | .c | math => code |
| 3 | convert C code to shared library | .so | code => machine code |
The optional file formats for ODEs are .vf, .mod, .ode, and plain text (.txt).
We generate C code in R for the SBtab model using the computer algebra package Ryacas, which interfaces between R and yacas – both need to be installed.
These three steps can be performed with the R commands below. SBtabVFGEN refers to our external package.
Convert SBtab to ODE
The following two commands are unrelated to code generation. They just find and load the biological model.
f <- uqsa::uqsa_example("AKAR4cl") # paths to the TSV files
sb <- SBtabVFGEN::sbtab_from_tsv(f) # loads the TSV filesWe convert the model into an ordinary differential equation (and create intermediate files, various formats).
odeModel <- SBtabVFGEN::sbtab_to_vfgen(sb)Convert the ODE to C code
The function generateCode below accepts the return value
of SBtabVFGEN::sbtab_to_vfgen() (a list of ode properties)
and returns a character vector of C code:
C <- uqsa::generateCode(odeModel)This returns the generated code as a character vector, it can be
viewed (and written to a file) using the cat function.
The print the first few lines of the generated code (25 lines in the
example), you can run the following command. You can use the
file= argument of cat to print it into a
file.:
The following command saves the C source code in the file “AKAR4cl_gvf.c”.
cat(C, sep = "\n", file = "AKAR4cl_gvf.c")The file just created contains the following functions:
grep AKAR4cl_ ./AKAR4cl_gvf.c
#> int AKAR4cl_vf(double t, const double y_[], double *f_, void *par){
#> int AKAR4cl_jac(double t, const double y_[], double *jac_, double *dfdt_, void *par){
#> int AKAR4cl_jacp(double t, const double y_[], double *jacp_, double *dfdt_, void *par){
#> int AKAR4cl_func(double t, const double y_[], double *func_, void *par){
#> int AKAR4cl_funcJac(double t, const double y_[], double *funcJac_, void *par){
#> int AKAR4cl_funcJacp(double t, const double y_[], double *funcJacp_, void *par){
#> int AKAR4cl_default(double t, double *p_){
#> int AKAR4cl_init(double t, double *y_, void *par){Convert C code to shared library
The following command compiles the model (if not yet compiled).
modelName <- checkModel("AKAR4cl","AKAR4cl_gvf.c")
#> building a shared library from c source, and using GSL odeiv2 as backend (pkg-config is used here).
#> cc -shared -fPIC `pkg-config --cflags gsl` -o './AKAR4cl.so' 'AKAR4cl_gvf.c' `pkg-config --libs gsl`(Optional) Generate code for deSolve (ODE solver in R):
It is also possible to create R code, intended for use with deSolve. Combining all previous function calls into one (writing to file immediately).
rFile <- paste0(comment(odeModel),".R")
cat(uqsa::generateRCode(odeModel),sep="\n",file=rFile)
source(rFile)
# example call:
p <- AKAR4cl_default(0.0)
y0 <- AKAR4cl_init(0.0,p)
print(y0)
#> AKAR4p C
#> 0 0A test-simulation with deSolve:
if (require(deSolve)){
t_ <- seq(0,50,length.out=128)
y_ <- ode(y0,t_,AKAR4cl_vf,p)
plot(t_,y_[,1],type='l',main="AKAR4cl",xlab="time")
}
#> Loading required package: deSolve
Simulate
We now show how to simulate the model the same conditions as in the experiments (e.g., same initial conditions). More details on simulating the model can be found in the page “Simulating a model”.
# import the experimental data
ex <- SBtabVFGEN::sbtab.data(sb,odeModel$conservationLaws)
## we create a closure called "sim" a function that internally remembers the "experiments" (ex)
sim <- simulator.c(ex,modelName) # ex holds the instructions for the solver
par <- as.matrix(sb$Parameter[["!DefaultValue"]]) # one column - but can be several sets (as columns)
## sim can be called with any parameter vector - or several and will always simulate the same set of experiments
sr <- sim(par) # simulation results (one per experiment)
stopifnot(length(sr) == length(ex))
i <- seq(70)
t <- ex[[1]]$outputTimes[i]
y <- ex[[1]]$outputValues[[1]][i]
dy <- ex[[1]]$errorValues[[1]][i]
z <- sr[[1]]$func[1,i,1] # third dimension is the par column (just one here)
plot(t,z,bty='n',type='l', ylim=c(90,200))
points(t,y)
arrows(t,y,t,y+dy,angle=90, length=0.01)
arrows(t,y,t,y-dy,angle=90, length=0.01)
legend("bottomright",legend=c("simulation","data"), lty=c(1,1), pch=c(NA,1))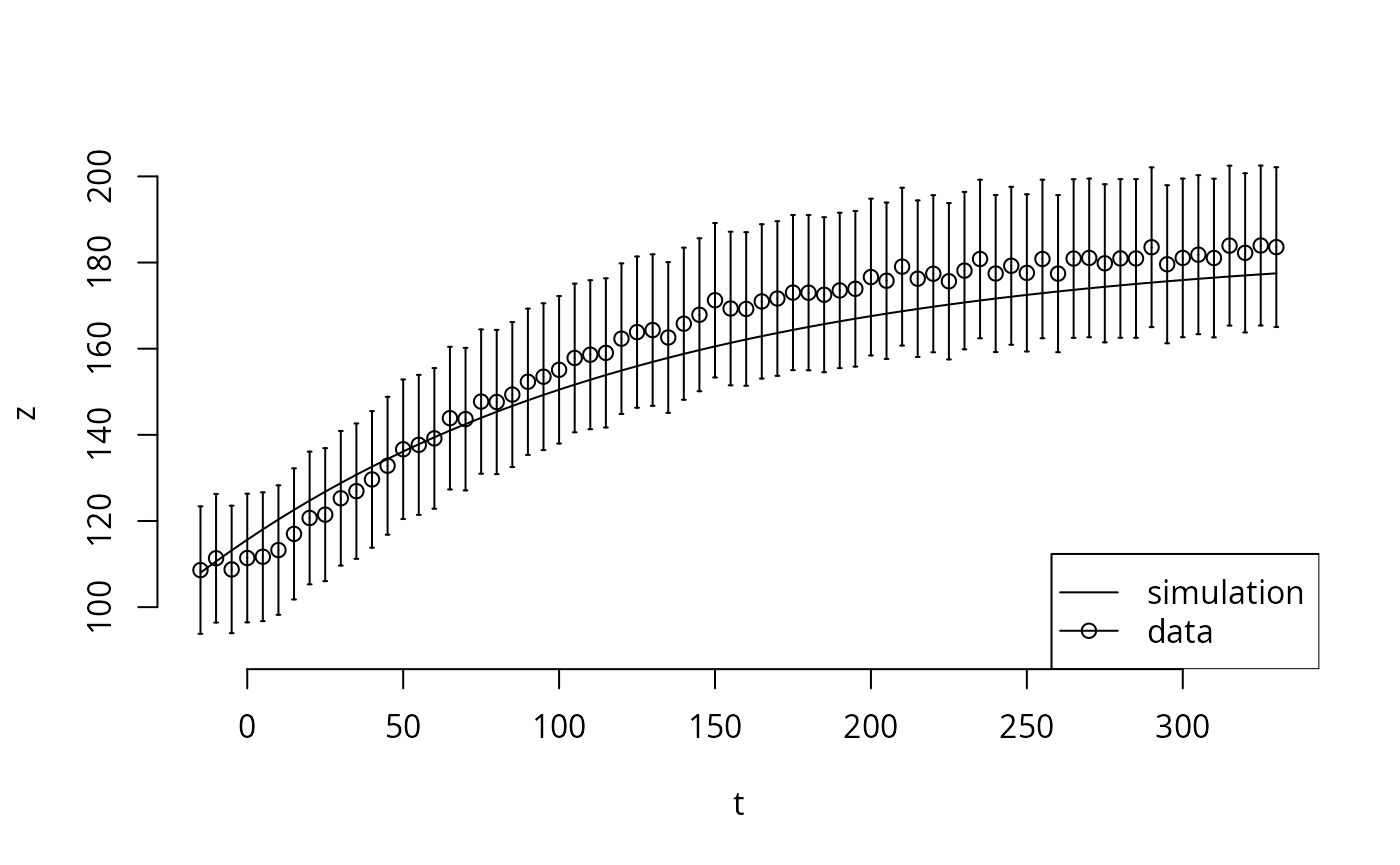
The simulation result sr is a list with several
components, including:
-
state, state variable trajectories -
func, output functions
The output functions usually correspond to the data (in some way), but may need some sort of normalization. In this example, the relationship is direct.
Both state and func have three indices:
sr[[l]]$state[i,j,k] # is a numberThis corresponds to experiment l, state variable
i, time-point j, and parameter column
k (relevant when par is a matrix with multiple
columns).
The previously created modelName variable has a comment
attribute (that you can also set yourself, there is no magic there).
This attribute indicates the location of the shared library used for
simulations. With this, ytou don’t have to re-make it every time - as
long as the model doesn’t change (i.e., checkModel does not
need to be re-called):
comment(modelName) <- "./AKAR4cl.so" # this is sufficient if the .so file already existsSimulations of Several Parameter Sets
The following commands allow you to simulate multiple parameters
(saved in the matrix P) at the same time.
t <- as.numeric(ex[[1]]$outputTimes)
np <- NROW(par)
REPS <- 50
P <- matrix(runif(np*REPS,min=0,max=as.numeric(par)*2),np,REPS)
dim(P)
#> [1] 3 50
stm <- Sys.time()
sr <- sim(P)
etm <- Sys.time()
difftime(etm,stm)
#> Time difference of 2.111909 secsLet’s plot all trajectories in one picture:
T <- rep(c(t,NA),REPS) # the NA value will break the line
Z <- as.numeric(sr[[1]]$func[1,c(seq_along(t),NA),]) # at the end, so it doesn't loop
plot(T,Z,type='l')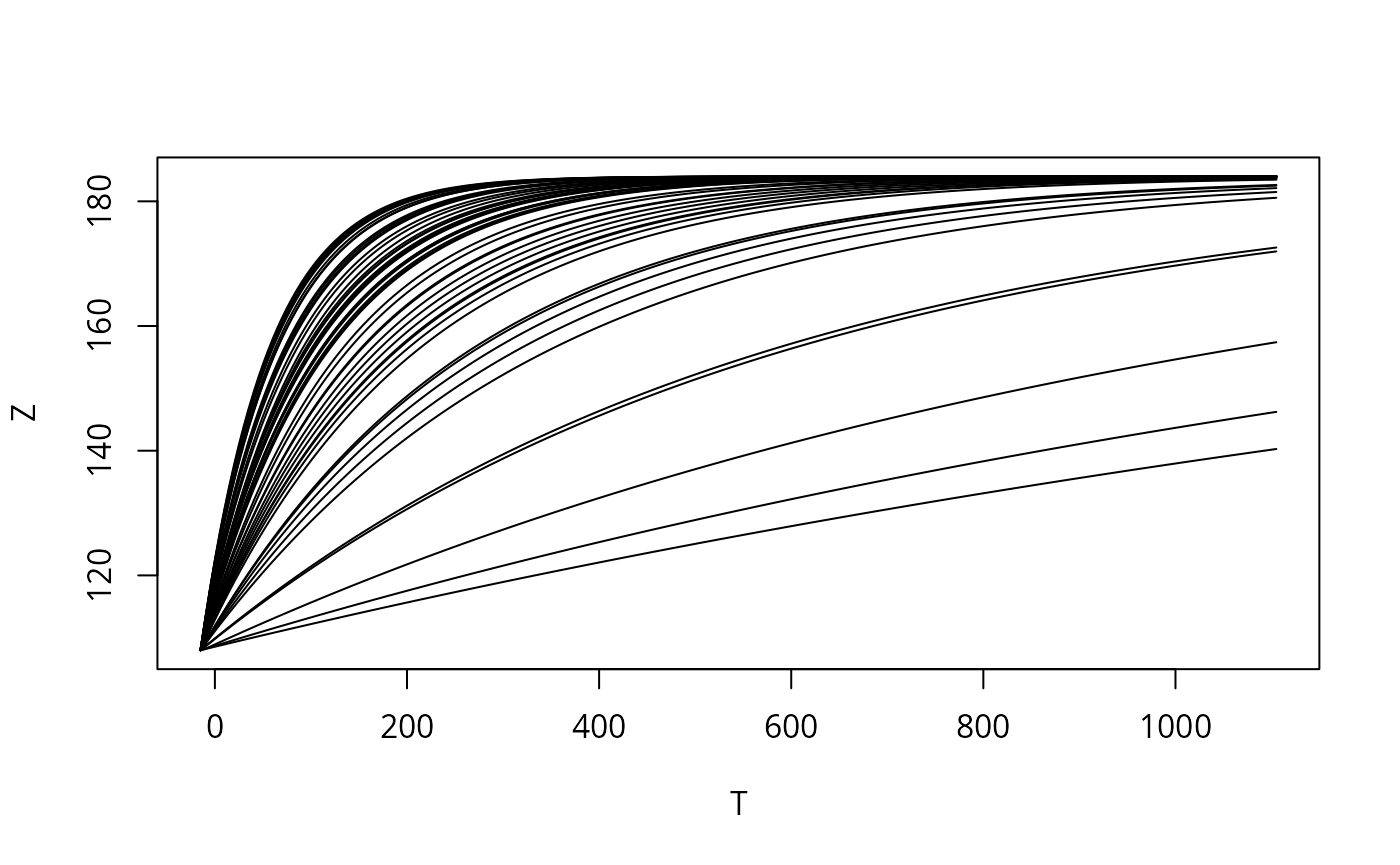
The new result is (just like the old one) an array with three indexes:
sr[[l]]$func[i,j,k] # a number-
ioutput function -
jtime index -
kselects the trajectories that belong toP[,k]
Clean-up (delete temporary files):
gf <- sprintf("AKAR4cl%s",c(".vf",".tar.gz",".zip",".so"))
print(gf)
#> [1] "AKAR4cl.vf" "AKAR4cl.tar.gz" "AKAR4cl.zip" "AKAR4cl.so"
file.remove(gf)
#> [1] TRUE TRUE TRUE TRUEOther Formats
The SBtab content can be a normal spreadsheet.
If it is a Google spreadsheet (which can be good for collaborative
work), you should write it until fairly satisfied with all components
and then export it as an .xlsx, or .ods file.
The gnumeric application can automatically convert both
.xlsx and .ods to a set of .tsv
files if you want the benefits of version control (which works on text
files). Both .ods and .xlsx work:
-
SBtabVFGEN::sbtab_from_odsfor ODS files -
SBtabVFGEN::sbtab_from_excelfor XLSX files
Our scripts find the tables by name:
- TSV files:
TableNameattribute is the name of the table - ODS files: the Sheet’s name is the name of the table,
TableNameattribute is not parsed - Excel files: the Sheet’s name (same as ODS)
Tab Separated Values
For more information on how to solve common tasks regarding tsv files, see the tsv topic. We obtain a list of all tsv files in the current directory, and then import the contents, like this:
modelFiles <- dir(pattern='[.]tsv$')
SBtab <- sbtab_from_tsv(modelFiles)Open Document Spreadsheet
These files can be created with Libre Office, Apple’s numbers program, gnumeric, and any web-hosted spreadsheet application (like Google spreadsheets).
modelFiles <- "DemoModel.ods"
SBtab <- sbtab_from_ods(modelFiles)Excel
These can be created with MS Excel, and loaded like this:
modelFiles <- "DemoModel.xlsx"
SBtab <- sbtab_from_excel(modelFiles)Parameters
The R code generated by ode.sh includes a list with the
same functions, but with generic function names:
-
model$vf()vector field -
model$jac()Jacobian -
model$init()initial values -
model$par()default parameters in linear scale- regardless of what the scale was in the SBtab file
- this function returns the long parameter vector (Parameter and Input)
Observe that model$par() returns the ODE model
parameters, which can be more than the biological parameters, the
components are:
- biological parameters (
SBtab$Parameter) - original input parameters (
SBtab$Input) - derived input parameters, from conservationlaws (conserved constants)
The inputs are allowed to be different for each experiment (by
definition), the biological parameters are the same for all experiments
simulated in one go. Therefore, we only supply the biological parameters
(the first n) to the simulator and it retrieves the others
from the experiments variable, by concatenation:
Usually, the number of leading parameters
(sb$Parameter[[“!DefaultValue”]]) does not change. This is all the
simulator needs as the inputs are already in ex.